Meditsiin, millele viidatakse kui Roberti sündroom tõsine, autosomaalselt retsessiivne pärilik väärareng. Roberts sündroomi nimetatakse mõnikord Appelt-Gerken-Lenzi sündroom, Pseudothalidomiidi sündroom ja ka Roberts SC föcomelia määratud. Need nimed ei kirjelda erinevaid etappe ega vorme, vaid põhinevad peamiselt sündroomi avastajatel.
Mis on Roberti sündroom?

© bluebackimage - stock.adobe.com
Nagu Roberti sündroom kirjeldab väga harva esinevat väärarengut, mis tekib geneetilise määramise või mutatsiooni tõttu. Eriti iseloomulik on nelja jäseme puudumine. Prognoosid on reeglina väga halvad; paljudel juhtudel on füüsiline või vaimne areng tõsiselt piiratud. Enamik kannatanutest sureb sünnituse ajal.
Lõpuks meenutavad Roberti sündroomi all kannatajad ka Contergani ohvreid, kuna neil on sarnased deformatsioonid ja väärarengud. Roberti sündroomi ennetamiseks ei ole põhjuslikku ravi ega ennetavaid meetmeid. Roberti sündroomi diagnoosinud arstid püüavad geneetilise testi põhjal peamiselt leevendada sümptomeid ja parandada haigete elukvaliteeti. Roberti sündroomi kirjeldati esmakordselt 1919. aastal.
Esimese teadusliku kirjelduse kirjutas ameerika kirurg John Bingham Roberts. Rohkem teadmisi oli aga vaja 67 aastat. See oli Hans Appelt, Widukind Lenz ja Hartmut Gerken, kolm saksa geneetikut, kes viisid oma esimese uuringu läbi 1966. aastal. Sel põhjusel nimetatakse pärilikku väärarengut Roberti sündroomiks või nimetatakse seda ka Appelt-Gerken-Lenzi sündroomiks.
põhjused
Roberti sündroom tekib ESCO2 geeni mutatsioonide tõttu. ESCO2 geen asub 8. kromosoomi geeni lookuses p21.1. Ennekõike mängib olulist rolli ESCO2 geeniprodukt; lõpuks on see niinimetatud N-atsetüültransferaas, mis koosneb inimestel täpselt 601 aminohappest.
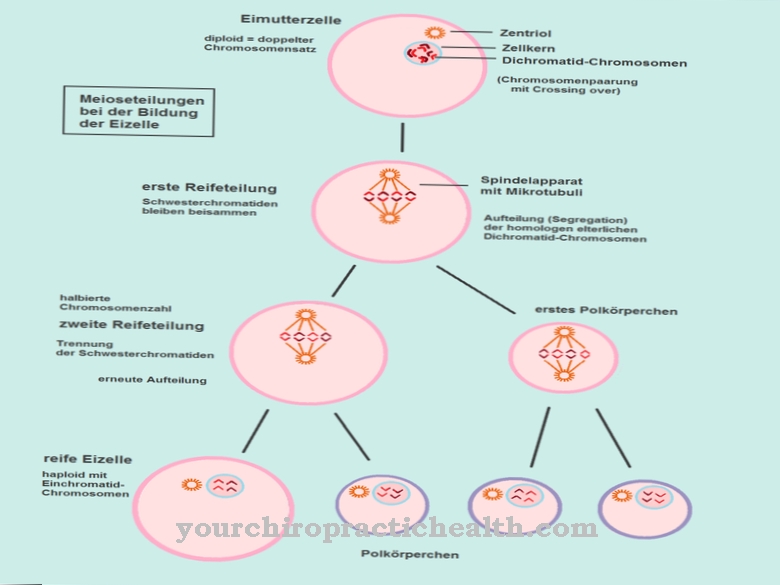
Kui S-faas siseneb ja rakkude jagunemine algab, kromatiidid kahekordistuvad. See lähenemine käivitab sündroomi. Siiani pole mutatsiooni kandjasagedus teada. ESCO2 geen koosneb kokku üheteistkümnest eksonist mõõtmetega 30,3 kb; puudub teave selle kohta, milline kandesagedus on olemas. Mutatsiooni põhjused pole teada.
Sümptomid, tervisehäired ja nähud
Mõjutatud inimestel, kes põevad Roberti sündroomi või kes ei ole sündinud surnult sündinud või surevad vahetult pärast sündi, ilmnevad arvukalt sümptomeid, mis näitavad vastavat mutatsiooni. Seal on vaimne alaareng, mikrotsefaalia (pea on väike), fokomelia (nn hülgejäseme) ja ka brahütsefaalia (lühike või ümar peavalu).
Peaaegu kõigil juhtudel on näha huulte ja suulaelõhe; Samuti on kliitori ja peenise hüperplaasia (laienemine). Haigestunute sarvkest on hägune või saab arst tuvastada ka siseorganite (südame või neerude) deformatsioone.
Diagnoos ja haiguse kulg
Arst paneb alguses kahtlustatava diagnoosi. Ta saab seda teha ilma probleemideta - lähtudes sümptomitest. Diagnoosi saab kinnitada alles siis, kui mutatsioon on tuvastatud geneetilise testi abil. Kuni pole geneetilisi tõendeid selle kohta, et Roberti sündroomi eest vastutav mutatsioon eksisteerib, diagnoositakse kahtlus, hoolimata sellest, kui selged on inimese sümptomid.
Paljudel juhtudel surevad kannatanud pärast sündi või on nad juba surnult sündinud. Kuid on ka üksikjuhtumeid, kus normaalne vaimne areng dokumenteeriti. Siiski tuleb mainida, et see on absoluutne erand. Haiguse käik ja prognoosid on enamasti negatiivsed.
Terapeutilised meetmed võivad teoreetiliselt vähendada haiguse kulgu või soodustada sümptomeid, kuid ka siin ei saa rääkida positiivsest käigust ega positiivsest prognoosist.
Tüsistused
Roberti sündroomi all kannatajad põevad tavaliselt sündides või vahetult pärast seda. Kui haige laps jääb ellu, kannatab ta peaaegu alati tõsiste vaimsete ja füüsiliste kahjustuste käes. Nelja jäseme puudumine ja muud tüüpilised kõrvalekalded on lapsele seotud märkimisväärse valuga. Vaimupuue on seotud mitmesuguste komplikatsioonidega - alates arenguhäiretest kuni konkreetsete sekundaarsete haiguste ja sotsiaalse tõrjutuseni.
Üldiselt kannatab kannatanud inimene mitmesuguste kaebuste ja nende hilise mõju tõttu. See mõjutab lapse ja vanemate vaimset seisundit. Normaalse vaimse arengu korral vajab kannatanud inimene kogu elu tuge. Muud tüsistused sõltuvad konkreetsetest sümptomitest.
Jäsemete puudumist seostatakse voodipuhkuse ja selle tüüpiliste tagajärgedega, huule ja suulaelõhe põhjustavad kõnehäireid. Üksikute kaebuste käsitlemisel võivad tekkida täiendavad komplikatsioonid. Infektsioonid ja närvivigastused tekivad sageli operatsiooni ajal.
Haigestunute üldiselt halva füüsilise seisundi tõttu ei saa välistada südame-veresoonkonna probleeme ja haavade paranemise häireid. Ravimite manustamisel võivad tekkida kõrvaltoimed ja koostoimed või tekkida allergilised reaktsioonid.
Millal peaksite arsti juurde minema?
Roberts'i sündroomi all kannatavad lapsed vajavad hoolikat ravi. Erinevaid füüsilisi ja vaimseid häireid ravitakse rekonstruktiivselt ja kosmeetiliselt, et parandada heaolu ja elukvaliteeti. Füsioteraapia on tavaliselt vajalik käte ja jalgade väärarengute kompenseerimiseks. Haigestunud laste vanemad peaksid arstiga rääkima vajalikest terapeutilistest abinõudest ja konsulteerima vastava spetsialistiga. Sõltuvalt sümptomitest osalevad ravis muu hulgas ortopeedid, neuroloogid, kirurgid, silmaarstid ja dermatoloogid.
Lastearst võib üle viia regulaarsed järelkontrollid, kui last ei pea ravima statsionaarina. Tavaliselt vajavad terapeutilist tuge ka kannatanud lapse vanemad ja sugulased. Kui laps on surnult sündinud või haigus on surmaga lõppenud, on see eriti suur koormus vanematele, kes peaksid juba varakult pöörduma sobiva psühholoogi poole, et trauma läbi saada ja sellest üle saada. Kuna Roberti sündroom on geneetiline haigus, on geneetiline nõustamine kasulik, kui soovite uuesti lapsi saada.
Teraapia ja ravi
Roberti sündroomi korral põhjuslikku ravi ei ole. See tähendab, et sümptomeid ravitakse peamiselt nii, et haigestunud inimese elukvaliteeti saaks võimalikult palju parandada. Siiski tuleb mainida, et paljudel juhtudel on väärarengud nii drastilised, et abi saab anda ainult vähesel määral.
Arst peab ise hindama, kui palju ravi võib aidata. Roberti sündroomi tuleb hinnata ainult individuaalselt, et seejärel otsustada, millist ravi saab aeg-ajalt läbi viia.
Ennekõike on olemas parandusmeetmed, et elukvaliteeti saaks parandada. Arst otsustab kirurgiliste paranduste üle, mis on peamiselt kosmeetilised ja funktsionaalsed. See annab võimaluse tõsta mõjutatud inimese elukvaliteeti. Üks probleemivabadest parandustest on huule ja suulae lõhe töötlemine.
Mõnikord võib nn käelõikuse sekkumine hõlbustada objektide hoidmist või haaramist. Kuna siseelundite väärarengud on iseloomulikud Roberti sündroomile, viiakse siin läbi individuaalne ravi. Arst pöörab tähelepanu elundite väärarengute ja deformatsioonide ulatusele, nii et siin viiakse läbi ka individuaalseid ravimeetodeid.
Arst peab otsuse langetama ka siis, kui Roberti sündroom on nii väljendunud, et mõjutatud isikul on mõnikord vaid paar tundi või päeva, et mitte alustada täiendavat ravi. Lõpuks ootab arst ainult patsiendi loomulikku surma.
ärahoidmine
Roberti sündroomi ei saa vältida. Seda seetõttu, et pole ka teada, miks mutatsioon toimub või kas leidub soodsaid tegureid.
Järelhooldus
Roberti sündroomi järelravi peab toimuma iga haigestunud inimese jaoks eraldi, kuna sümptomid võivad olla väga erinevad. Tõsiste väärarengute korral ei saa patsienti aidata, nad surevad sageli sünnituse ajal või vahetult pärast seda. Järelhooldus hõlmab seejärel uimastiravi või palliatiivset ravi.
Kui on vaja operatsiooni, on kõige olulisemad abinõud haava hea hooldus ja kirurgilise armi kontrollimine. Sageli kaasneb sellega füsioteraapia. Järelhooldus võib hõlmata ka spetsialistide täiendavaid uuringuid, sõltuvalt sümptomitest, mis võivad ulatuda sarvkesta läbipaistmatusest siseorganite väärarengute tekkeni.
Sõltumatut järelravi tavaliselt ei toimu, kuna Roberti sündroom on krooniline haigus ja haigeid tuleb ravida kogu elu. Vastutav spetsialist peab koos sugulastega otsustama, millised ravivõimalused on pikas perspektiivis võimalikud. Võimalik, et selles osas tuleb konsulteerida ka psühholoogidega, kuna haigus võib olla asjaosalistele suur emotsionaalne koormus. Emotsionaalset tuge vajavad eriti lapse vanemad, kuna enamasti surevad patsiendid vahetult pärast sündi või on surnult sündinud.
Saate seda ise teha
Roberti sündroom võib esineda erineval kujul, mis võib võtta erinevaid kursusi. Sõltuvalt ravikuurist tuleb koos arstiga välja töötada individuaalne teraapia, mis hõlmab ka sugulaste eneseabimeetmeid.
Kerge haiguse korral vajavad haiged esmalt mitmesuguseid operatsioone ja füsioterapeutilist tuge. Enamasti on tegemist kõigi nelja jäseme väärarenguga, mistõttu on abivahendite, näiteks karkude või ratastooli kasutamine igal juhul vajalik. Sugulased peaksid igapäevaelus abi saamiseks tugikeskusega juba varakult ühendust võtma. Kuna kannatused on sugulaste jaoks märkimisväärne emotsionaalne koormus, on terapeutiline ravi kasulik ka vanematele.
Pärast operatsiooni tuleb last pidevalt jälgida. Isegi pärast haiglas viibimist on suurenenud komplikatsioonide oht. Vanemad peaksid ühendust võtma ka erikoolide ja erilasteaedadega. Mida varem neid abinõusid rakendatakse, seda vähem stressirohke elu võib olla haige lapsega. Roberts'i sündroomiga lapsed saavad kodus füsioteraapiat teha. Igal juhul peavad nad järgima tervislikke eluviise, piisava liikumise ja sobiva toitumisega.

.jpg)