Jouberti sündroom iseloomustab nii ajutüve kaasasündinud väärareng kui ka agenees (inhibeerivad väärarengud, kinnituse puudumine, näiteks ajuvardad, lisa). Võib esineda ka tserebellide usa hüpoplaasia (vähearenenud). Patsientidel, kes kannatavad selle autosomaalse retsessiivse geneetilise defekti all, on muu hulgas ebanormaalne hingamiskäitumine ja ataksia.
Mis on Jouberti sündroom?

© Sashkin - stock.adobe.com
Inimesed, kellel on Jouberti sündroom kannatavad kesknärvisüsteemi arenguhäirete ja neist tulenevate funktsionaalsete häirete all. Meditsiinilised uuringud on vaieldavad selles osas, kas seda geneetilist häiret tuleks liigitada omaette haiguseks.
Haigestunud patsientidel on mitmesuguseid erinevaid sümptomeid. Seetõttu on lõplik diagnoosimine keeruline. JB-d iseloomustab ulatuslik geen lookuse heterogeensus. Siiani on tuvastatud mitu geenimutatsiooni. Mutatsioonide analüüs on väga ulatuslik.
põhjused
Jouberti sündroom kuulub primaarsete tsilofaatide rühma. Selle primaarse tsiliaadi või basaalkeha geneetilise häirega võivad tekkida erinevat tüüpi arenguhäired. Spetsiaalsete rakuprotsessidena täidavad cilia mitmesuguseid ülesandeid. Need toimivad keemia-, mehano- ja osmoosianduritena ning osalevad paljudes signaalimisteedes. Lisaks tagavad nad elundi normaalse arengu.
Nad säilitavad põhiliste arenguprotsesside kudede homeostaasi. Suur osa kaasatud valkudest moodustab interaktsiooni kaudu keeruka võrgu. Kui lisaks peamistele sümptomitele on mõjutatud ka muid organeid, on tegemist JSRD-ga (Jouberti sündroomiga seotud häire). Seda sekundaarset haigust iseloomustavad täiendavad elundite manifestatsioonid, mis hõlmavad neere, maksa ja silmi.
See on geneetiliselt heterogeenne sündroom. Arstid on leidnud väärarenguid geenis NPHP6 / CEP290 (kodeerivad nefotsüstiini-6) või geenis NPHP8 / RPGRIP1L (kodeerivad nefotsüstiin-8). Muud geenimutatsioonid on MKS3, ARL13B, AHI1, CC2DA2, TMEM216 ja INPP5E. Ainult vähestel patsientidel on NPHP4 ja NPHP1 mutatsioonid.
Sümptomid, tervisehäired ja nähud
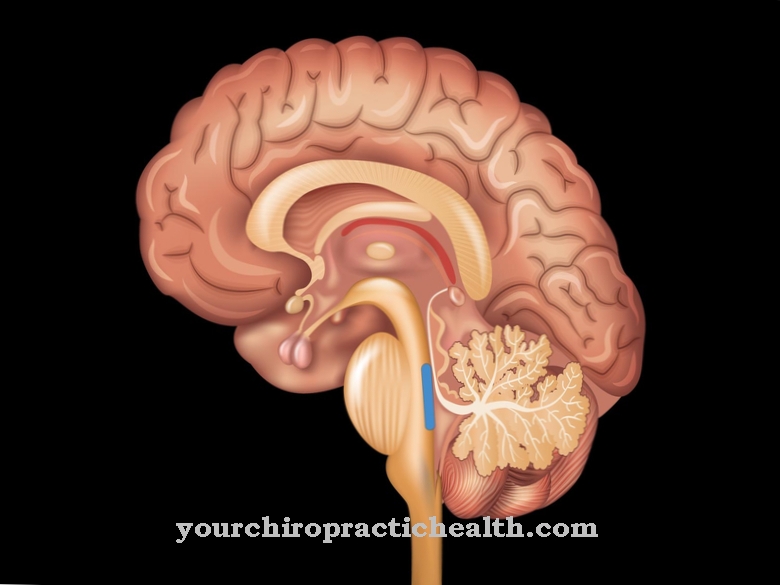
Patognomooniline tunnus on "molaarsete hammaste märk" (MTS), mida saab kindlaks teha "aksiaalse T1-ga kaalutud aju magnetresonantstomograafia" abil. Seda tunnust iseloomustab väikeaju või väikeaju usu agenees või hüpoplaasia. Lisaks on tagumine ristsuunaline fossa (aju jalgade vaheline auk) tugevalt sisse tõmmatud ja väikeaju vartel on keskmise aju väärarengu tõttu silmatorkav parem kuju.
Lisaks MTS-le kannatavad patsiendid sageli hingamisteede häirete, ataksia, lihaste hüpotensiooni ja psühhomotoorse alaarengu all. 8–19 protsendil haigestunutest ilmneb postaksiaalne polüdaktiliselt (mitu sõrme) ja kuuel protsendil kuklakujuline (meningo) entsefalocele, kus aju tagumine osa on punnis.
See deformatsioon registreeriti esmakordselt 1969. aastal. Levimus on umbes 1: 100 000, suhe näitab, kui harva haigus ilmneb. Pärast esimest meditsiinilist ülevaatust on dokumenteeritud vaid sada juhtumit. Kuna see geneetiline defekt ilmneb erinevates vormides ja variantides, eeldavad arstid geneetikas mitmeid muutusi.
Täpset kõrvalekallet ei ole veel lõplikult kinnitatud. X-kromosoomi mutatsiooni peetakse siiski kindlaks. See häire kandub edasi autosomaalse retsessiivse pärimise alusel. Puudub vermis tserebelli (väikeaju, väikeaju uss), võrkkesta kahjustus ja märgatav iiris.
Vastsündinu perioodil sageli esinevad sümptomid ja kaebused on nüstagm ja ebaregulaarne hingamismuster, nagu episoodiline tahhüpnea ja apnoe. Väikestel lastel võib tekkida hüpotoonia. Vananedes areneb tasakaal ja ebaühtlane kõnnak (ataksia). Neid peamisi sümptomeid tuntakse ka kui motoorseid verstaposte.
Patsientidel on erinev kognitiivsete võimete tase ja nad võivad olla tõsiselt kahjustatud, kuid nad võivad näidata ka normaalset intelligentsustaset. Võimalik on ka oculo-motoriline apraksia (liikumishäire).
Sellele geneetilisele defektile on iseloomulikud kraniofaciaalsed kõrvalekalded, nagu suur pea, ümarad ja kõrged kulmud, silmatorkav (väljaulatuv) otsmik, deformeerunud suu, rütmiliselt liikuv ja väljaulatuv keel ning sügavalt asetsevad kõrvad. Aeg-ajalt on sümptomiteks nefrofütis, võrkkesta düstroofia ja polüdaktiilsus.
Diagnoos ja haiguse kulg
Diagnoosimisel võetakse aluseks eelnevalt viidatud ataksia, hüpotensiooni, okulomotoorse apraksia, avatud vermis cerebelli iseloomulikud verstapostid pärast 18. rasedusnädalat ja arengu hilinemine. Lisaks tehakse MRT-s iseloomulik neuroradioloogiline leid - MTS (Molaarne hammaste märk).
See molaarmärgiks tuntud omadus on tingitud pastilli ja kesk aju väärarengutest, samuti väikese aju ussi hüpoplaasiast. Diferentsiaaldiagnoosid põhinevad JS-iga tihedalt seotud haigustel, näiteks JSRD-l (Jouberti sündroomiga seotud häire), Dandy-Walkeri väärarendil (väärarenguga tserebraalne uss ilma MTS-ita), okulomotoorse apraksia tüüpidel 1 ja 2, ponto-tserebraalse hüpoplaasia ja atroofiaga, 3-c Sündroom, orofatsio-digitaalsed sündroomid II ja III, samuti Meckel-Gruberi sündroom.
I etapp hõlmab geenide JBTS5 (53 kodeerivat eksonit), JBTS3 (26 kodeerivat eksonit), JBTS6 (28 kodeerivat eksonit) ja JBTS9 (36 kodeerivat eksonit) järgmise põlvkonna järjestuspõhist paneelianalüüsi. JBTS4 geeni testitakse homosügootse deletsiooni suhtes multipleksse PCR abil. II etapis analüüsitakse teisi JB geene PCR-ga (protsess, mis dubleerib DNA ahelas geenijärjestusi sõltuvalt ensüümist) ja järgneva Sangeri sekveneerimisega, sõltuvalt fenotüübilistest tunnustest, mis vastab mutatsioonide sageduse vähenemisele.
Kromosoomide tasakaalustamatuse välistamiseks viiakse läbi diferentsiaaldiagnostika SNP-i massiivi analüüs. Üksmeele või kui perekonnas on teada mitu haiget isikut, viivad arstid läbi homosügootsuse sõeluuringu geeni külgnevas mikrosatelliidimarkeris sidumisanalüüsi ja järgneva geenianalüüsi abil, kasutades Sangeri järjestust. Diagnostilise materjalina võetakse lastelt kaks kuni kümme milliliitrit EDTA verd, täiskasvanutest - viis kuni kümme milliliitrit.
Samuti sobib DNA või koematerjal. I etapp: genoomse DNA materjali uuritakse duplikatsioonide või deletsioonide olemasolu suhtes NPHP1 geeni kvantitatiivse analüüsi abil, kasutades MLPA. Genoomis uuritakse väga väikestes DNA kogustes üksikute eksonite (geenisegmentide) deletsiooni ja dubleerimist. II etapp: Seni tuvastatud geenide kodeeritud eksoone hinnatakse järgmise põlvkonna sageduste abil. Splitseerimise kohti rikastatakse sondi hübridiseerimisega.
Tüsistused
Jouberti sündroom põhjustab enamiku patsientide mitmesuguseid vaevusi. Tavaliselt põhjustab see lühikest kasvu, hingamishäireid ja lisaks alaarengut. Samuti saab piirata lapse vaimset arengut. Hingamisraskused võivad põhjustada ka õhupuudust, mida tuleb kindlasti ravida.
Pole harvad juhud, kui inimese vanemad kannatavad raske depressiooni või muude psühholoogiliste häirete all. Patsientidel esinevad ka tasakaaluhäired ja sageli on nad piiratud liikumisvõimega. Silmadele ja kõrvadele põhjustatud ebamugavustunne, mis põhjustab kuulmislangust või nägemisprobleeme, ei ole haruldane. Jouberti sündroom halvendab oluliselt patsiendi elukvaliteeti.
Erinevate ravimeetodite abil saab Jouberti sündroomi piirata ja ravida. Kahjuks ei saa põhjuslikku ravi läbi viia. Hädaolukordades saab õhupuuduse korral läbi viia ka hädaolukorras ventilatsiooni. Ravis endas pole mingeid erilisi tüsistusi. Üldiselt ei saa ennustada, kas Jouberti sündroom lühendab patsiendi eeldatavat eluiga.
Millal peaksite arsti juurde minema?
Rase ema peaks raseduse ajal osalema kõigis saadaolevates kontrollides. Uurimisel kontrollitakse nii raseda kui ka sündimata lapse tervislikku seisundit. Kuna Jouberti sündroomi saab diagnoosida juba 18. rasedusnädalal, on soovitatav kasutada tervisekindlustusseltside soovitatud ennetavaid tervisekontrolle. Lisaks, kui vanemate esivanemate ajaloos on geneetiline defekt, on üldjuhul soovitatav geneetiline nõustamine ja uurimine.
Kui ebatõenäoline, et emakas ei avastatud mingeid rikkumisi, toimuvad sünnitusarstide ja lastearstide automaatsed kontrollid kohe pärast sünnitust. Nende uuringute käigus saab tuvastada hingamishäireid. Kui lapse vanemad märkavad ebaharilikke lahknevusi, mis on varem märkamata jäänud, tuleks tähelepanekuid arstiga arutada. Füüsiliste iseärasuste, lühikese kehaehituse või deformatsioonide ilmnemisel tuleb pöörduda arsti poole.
Kui võrrelda samaealiste lastega keeleprobleeme või vaimset alaarengut, tuleb pöörduda arsti poole. Põhjuse väljaselgitamiseks on vaja uurimisi. Mida varem diagnoos tehakse, seda varem saab lapse toetamiseks alustada suunatud ravimeetodeid. Seetõttu tuleks kõrvalekalde esimeste märkide korral konsulteerida arstiga.
Ravi ja teraapia
Vanematel on õigus geeninõustamisele. Ravivõimalused on sama mitmekesised, kuna selle haiguse põhjused on mitmekesised. Motoorse arengu häirete ja hüpotensiooni korral tulevad mängu hariduslikud tugiprogrammid, keele-, töö- ja tegevusteraapia, millel võib olla haiguse kulgemisele kasulik mõju.
Ebanormaalsete hingamisharjumustega inimestele võib anda ka hapnikuasenduse või ventilatsiooni. Kergete sümptomitega patsientidel on positiivne prognoos. Tõsiselt kannatanud patsientide eest peab hoolitsema ekspertide tugikeskus.
Outlook ja prognoos
Jouberti sündroomi prognoos on kehv. See sündroom on geneetiline haigus. Praeguste meditsiiniliste, teaduslike ja juriidiliste nõuete kohaselt ei saa seda ravida. Teadlastel ja arstidel pole seaduslikult lubatud sekkumiste kaudu inimese geneetilisi seisundeid muuta. Sel põhjusel on ravi suunatud selliste ravimeetodite kasutamisele, mille eesmärk on olemasoleva elukvaliteedi parandamine. Ilma arstiabi kasutamata väheneb veelgi patsiendi heaolu.
Mida varem saab sündroomi diagnoosida ja ravida, seda paremad on tulemused. Hädaolukordades on vaja asjaomase inimese hädaabiventilatsiooni, vastasel juhul võib patsient enneaegselt surra. Ehkki arvukalt ravimeetodeid on kokku pandud ja neid rakendatakse individuaalses raviplaanis, võib olemasolev haigus viia sekundaarsete häireteni. Need halvendavad üldist prognoosi.
Olemasolevad funktsionaalsed häired või muud liikumispiirangud võivad põhjustada vaimuhaigusi. Ajutine või püsiv depressioon, meeleolumuutused või isiksuse muutused on paljudel patsientidel dokumenteeritud. See kujutab endast asjassepuutuvale inimesele ja keskkonnale täiendavat koormust. Jouberti sündroomiga patsiendi igapäevaelu saab sageli hallata ainult sugulaste piisava abi ja toetuse abil. Tasakaaluhäired ja ataksia muutuvad vanusega raskemaks.
ärahoidmine
Kuna täpset geneetilist põhjuslikku seost ei ole veel lõplikult kindlaks tehtud, pole kliinilises mõttes ennetavaid meetmeid. Ainus viis inimorganismi väärarengute vastu on tervisliku eluviisi järgimine.
Järelhooldus
Enamikul juhtudest pole Jouberti sündroomiga patsiendil otseseid ega spetsiaalseid jälgimisvõimalusi, nii et haigestunud inimene sõltub peamiselt haiguse kiirest ja ennekõike varast diagnoosimisest. Mida varem haigus tuvastatakse, seda parem on edasine kulg. Seetõttu on esimeste sümptomite ja nähtude ilmnemisel soovitatav pöörduda arsti poole.
Selle haigusega sõltub haigestunud inimene tavaliselt intensiivravist ja teraapiast, mis võib sümptomeid leevendada. Vanemate ja lähisugulaste abi ja tuge on samuti väga vaja, et kannatanud isik saaks võimalikult normaalset elu elada. Sageli saab füsioteraapia või füsioteraapia harjutusi läbi viia ka oma kodus, mis võib sümptomeid leevendada.
Sümptomeid ei saa alati täielikult leevendada. Kontaktid teiste Jouberti sündroomi all kannatajatega võivad samuti olla väga kasulikud, kuna teabe vahetamine pole haruldane. Reeglina ei vähenda see haigus haigestunud inimese eluiga.
Saate seda ise teha
Jouberti sündroom on ravimatu ja ka igapäevane abi on keeruline. Kaasasündinud haiguse sümptomid on enamikul juhtudel vältimatud. Siiski on võimalik, et mõnda neist leevendatakse.
Kuna hingamine on mõjutatud isikutel eriti häiritud, on see lähtepunkt. Abiks võib olla optimeeritud ruumikliima. Kuiv kütteõhk võib hingamisprobleeme veelgi süvendada. Sama mõju avaldab ka liiga külm õhk. Ideaalis on ruumi temperatuur umbes 20 ° C ja õhuniiskus umbes 50 protsenti. Eelkõige toataimed võivad optimaalse sisekliima soodustada. Teise võimalusena võib ruumi paigutada ka niisked rätikud, et hoida õhuniiskus soovitud tasemel. Sisekliimat saab jälgida hügromeetri abil. Teine lähtepunkt, mis on suunatud ka hingamisele, on hingamisharjutused. Regulaarne kasutamine parandab muidu automaatse protsessi tajumist. Sel viisil saate ära hoida liiga kiire hingamise ja hingamise pausi.
Samuti on mõistlik, kui kannatanud ei maga toas üksi. Sugulased võivad une ajal märgata hingamise pause ja äratada patsienti või stimuleerida teda hingama. Kuid see on ainult ettevaatusabinõu.

.jpg)