Juures Saethre-Chotzeni sündroom on kraniosünostoosiga seotud haigus. Saethre-Chotzeni sündroom on kaasasündinud, kuna selle põhjused on olemuselt geneetilised. Haigust tuntakse lühendiga SCS. Saethre-Chotzeni sündroomi peamised sümptomid on koronaarõmbluse sünostoos ühel või mõlemal küljel, ptoos, asümmeetriline nägu, ebaharilikult väikesed kõrvad ja strabismus.
Mis on Saethre-Chotzeni sündroom?

© Matthieu - stock.adobe.com
Saethre-Chotzeni sündroom esineb suhteliselt harva. Arvestades sagedust 1: 25 000 kuni 1: 50 000, esineb Saethre-Chotzeni sündroom mõnevõrra sagedamini kui paljud muud pärilikud haigused. Kraniofaciaalsed väärarengud on tüüpilised Saethre-Chotzeni sündroomile. Kraniosünostoos toimub koos sümfalangismi ja sündaktialiatega.
Mõned arstid viitavad Saethre-Chotzeni sündroomile sünonüümidena Chotzeni sündroom või III tüüpi akrotsefalosündaktiliselt sündroom määratud. Põhimõtteliselt ulatub Saethre-Chotzeni sündroomi üldnimetus tagasi nende inimeste juurde, kes kirjeldasid haigust esmakordselt teaduslikult 1931. aastal. Need on kaks arsti Chotzen ja Saethre.
Lisaks on olemas spetsiaalne Saethre-Chotzeni sündroomi vorm, milles patsientidel on lisaks tüüpilistele sümptomitele ka anomaaliaid silmalaugudel. Seda eripära nimetatakse terminiga Robinow-Sorauf sündroom. Selle haiguse osana on patsiendi suured varbad lõhenenud või mõnikord kahekordsed. Siiani on arvatud, et Robinow-Soraufi sündroom on Saethre-Chotzeni sündroomi alleelne variatsioon.
põhjused
Saethre-Chotzeni sündroomi patogeneesi põhjustavad tegurid on geneetilised defektid. Saethre-Chotzeni sündroomi arengu osas on siin eriti olulised nii deletsioonid kui ka punktmutatsioonid. Need rikkis protsessid toimuvad nn TWIST1 geenil. Spetsiaalne transkriptsioonifaktor kodeeritakse vastavas lookuses. See määratleb rakkude read ja on seotud diferentseerumisega.
Selle geeni geneetiliste defektide tõttu sulanduvad kolju õmblused liiga vara. Kustutuste tagajärjel ilmnevad mõjutatud patsiendid keskmisest märkimisväärselt. Lisaks on mõnel juhul nad vastutavad selle eest, et mõned Saethre-Chotzeni sündroomiga inimesed kannatavad vaimsete võimete all.
Sümptomid, tervisehäired ja nähud
Saethre-Chotzeni sündroomi kliinilised sümptomid on üksikjuhtudel erinevad. Tavaliselt on vastsündinutena mõjutatud inimestel sünostoos, mis tekib pärgarteri õmbluse piirkonnas. Seda õmblust nimetatakse ka lambdaõmbluseks või noolõmbluseks.
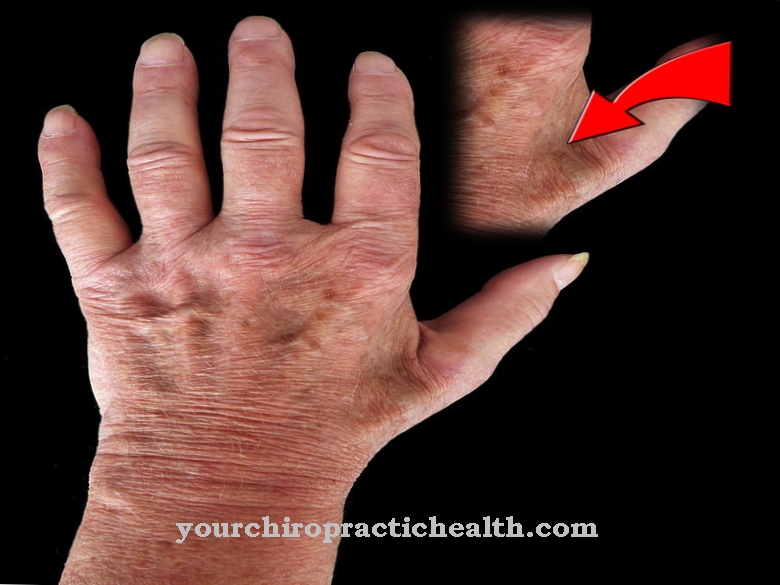
Seetõttu on patsiendi kolju ebahariliku kujuga. Lisaks on inimese silmis sageli selged asümmeetriad. Lisaks on patsientidel tavaliselt väga kõrge otsmik, sügava juuksejoone, väikeste kõrvade ja strabismiga. Levinud on ka ptoos, brahüdaktiliselt ja ninakanalite stenoos.
Keskmine ja nimetissõrm on naha piirkonnas sageli osaliselt üksteisega ühendatud. Enamikul juhtudest on Saethre-Chotzeni sündroomiga inimestel keskmine intelligentsuse jagatis. Mõnel neist mõjutatud isikutest ilmnevad intellektuaalses arengus siiski märkimisväärsed viivitused.
Mõnel Saethre-Chotzeni sündroomiga inimesel tekib kuulmislangus lapseeas. Sageli on see juhtiv kuulmislangus. Harvadel juhtudel kannatavad patsiendid lisaks üldistele sümptomitele ka lühikese kehaehituse, hüpoplastilise ülemise lõualuu luude, südame deformatsioonide või hüpertelorismi all.
Lisaks ilmneb mõnel patsiendil suulaelõhe, selgroolülide väärarengud, uvula bifida ja obstruktiivne uneapnoe. Kui sünostoos on eriti väljendunud, võivad mõjutatud patsiendid kogeda nägemise halvenemist, kolju suurenenud siserõhu tõttu pea piirkonnas valu ja epilepsiahooge. Äärmuslikel juhtudel surevad inimesed nende sümptomite tagajärjel.
Diagnoos ja haiguse kulg
Saethre-Chotzeni sündroomi diagnoosib tavaliselt eriarst. Perearst või lastearst suunab patsiendi vastava eriarsti juurde. Esimene anamnees enamasti lastega patsientide hooldusõiguse juuresolekul annab teavet praeguste sümptomite kohta.
Kliiniline läbivaatus põhineb tüüpilistel sümptomitel, mis mõnel juhul osutavad juba tugevalt Saethre-Chotzeni sündroomile. Tähelepanu keskmes on fenotüübi ja mitmesuguste kujutamismeetodite visuaalsed uuringud. Raviarst kasutab näiteks röntgen- ja CT-tehnikat. Need keskenduvad tavaliselt kolju, jäsemete ja selgroo piirkonnale.
Saethre-Chotzeni sündroomi kindel diagnoosimine on lõpuks võimalik geneetilise labori testi abil. Kui vastutavas geenis tuvastatakse deletsioon või punktmutatsioon, peetakse Saethre-Chotzeni sündroomi diagnoosi kinnitatuks. Olulist rolli mängib ka diferentsiaaldiagnostika, kuna mõned Saethre-Chotzeni sündroomi sümptomid on mõnikord segamini. Arst välistab Crouzoni sündroomi, Baller-Geroldi sündroomi, Pfeifferi sündroomi ja Muenke sündroomi.
Tüsistused
Saethre-Chotzeni sündroomi tagajärjel võib tekkida mitmeid tüsistusi. Tavaliselt kannatavad need, kelle intellektuaalne areng viibib märkimisväärselt. See võib viia sotsiaalse tõrjutuse ja selle tagajärjel psühholoogiliste komplikatsioonide, näiteks depressiooni või alaväärsuskomplekside tekkeni. Kuulmiskaotuse tagajärjel võivad igapäevaelus kehtida täiendavad piirangud ja kannatada saanud inimeste elukvaliteet halveneb.
Südame deformatsioonide ilmnemisel võib pikaajalise tagajärjena tekkida südamepuudulikkus. Lisaks sellele esinevad haiguse käigus korduvalt epilepsiahoogud, mis suurendavad õnnetuste riski. Halvimal juhul surevad mõjutatud isikud mitmesuguste kaebuste tagajärgede tõttu. Isegi positiivse tulemuse korral tekivad paratamatult füüsilised ja vaimsed komplikatsioonid. Üksikute väärarengute kirurgiline ravi on alati seotud riskidega.
Operatsiooni ajal võivad tekkida veritsus ja infektsioonid, samuti närvivigastused. See võib põhjustada haavade paranemise häireid, põletikku ja armistumist. Kirurgilise raviga kaasnevad ravimid peavad olema täpselt kohandatud patsiendi seisundile. Vastasel korral võivad mitmesuguste vaimsete ja füüsiliste häirete tõttu tekkida eluohtlikud kõrvaltoimed ja koostoimed.
Millal peaksite arsti juurde minema?
Saethre-Chotzeni sündroomi peab alati uurima ja ravima arst. Ravimata jätmisel võib see põhjustada tõsiseid tüsistusi, mis piiravad tõsiselt kannatanud inimese elu. Kuna see sündroom on geneetiline haigus, ei saa seda ka täielikult ravida. Seetõttu sõltub patsient puhtalt sümptomaatilisest ravist. Kui asjaomane isik soovib lapsi saada, võib sündroomi lastele edasikandumise vältimiseks läbi viia ka geneetilist nõustamist.
Kui patsient kannatab oluliselt hilinenud arengu all, tuleb pöörduda arsti poole. Ennekõike on intellektuaalne areng tõsiselt piiratud. Kuulmislangus või lühike iive võib näidata ka Saethre-Chotzeni sündroomi ja seda peaks alati uurima arst. Enamikul juhtudest ilmnevad ka südame deformatsioonid, nii et asjaomane inimene sõltub regulaarsetest südameuuringutest.
Saethre-Chotzeni sündroomi esialgse uurimise ja diagnoosimise võib läbi viia üldarst või lastearst. Edasist ravi viivad läbi erinevad spetsialistid ja see sõltub väärarengute tõsidusest.
Ravi ja teraapia
Saethre-Chotzeni sündroomiga patsiendid läbivad juba lapseeas mitmesuguseid kirurgilisi sekkumisi. Tavaliselt kasutatakse kranioplastikat. Kitsenenud hingamisteed on laiendatud ja suulaelõhe suletud. Regulaarsed kontrollid jälgivad kuulmist ning intellekti ja motoorseid arenguid. Samuti on oluline õigeaegselt ära tunda võimalik amblüoopia. Sama kehtib ka kroonilise kuluga papillaarse ödeemi kohta.
ärahoidmine
Praeguse uurimistöö seisu kohaselt ei saa Saethre-Chotzeni sündroomi ära hoida.
Järelhooldus
Saethre-Chotzeni sündroomi (SCS) järelmeetmete ulatus põhineb haiguse sümptomite iseloomul ja nende raskusastmel. Sümptomaatiliselt võib SCS põhjustada pea, keskosa, selgroo, jäsemete ja liigeste deformatsioone. Enamikul haigusjuhtudest ei arene eelnimetatud kehaosad korralikult välja. Deformatsiooni võivad jälgida tõsised sekundaarsed haigused.
Järelravi ülesanne on võimalikult varakult tuvastada ja ravida sekundaarsed SCS-i haigused. Järelmeetmete keskmes on haiguse kulgu jälgimine. Näiteks peapiirkonna deformatsioonid muutuvad tavaliselt kirurgiliselt. Seega saab asjaomase isiku pimedust vältida. Ümberkujundamise järelravi ajal uurib silmaarst mõjutatud inimese nägemisnärve igal kvartalil kuni 12. eluaastani.
Järelkontrollide eesmärk on võimalikult varakult välja selgitada, kas ja mil määral on kolju jälle liiga kitsas ja kui see on asjakohane, on olemas pimedaksjäämise oht. Kolju ümberkujundamisel võivad üksikjuhtudel tekkida epilepsiahoogud. Neid saab järelravi ajal ravida ravimitega.
Haiguse progresseerumisel saab epilepsia arengut neuroloogiliselt hoolikalt jälgida, kasutades elektroentsefalograafilisi salvestusi (EEG). Ülejäänud nelja kehaosa deformatsioonid põhjustavad regulaarselt sümptomaatilist piiratud liikuvust. Järelhooldus keskendub põhiliselt liikuvuse suurendamisele füsioteraapia abil. Valulikke kehaosi ravitakse ravimitega.
Saate seda ise teha
Eneseabi võimalused Saethre-Chotzeni sündroomi korral on suunatud haiguse optimaalsele käitlemisele. Patsient ja tema lähedased puutuvad kokku mitmesuguste toimetulekuprobleemidega, milleks tuleks neile võimalikult ulatuslikult valmistuda.
Patsiendi enesekindlus tuleb üles ehitada juba varakult. Nägemishäirete tõttu esindab häirimine igapäevaelus erilisi raskusi, millega patsiendil peaks olema võimalik emotsionaalselt silmitsi seista. Selleks võib kasutada ka psühhoterapeudi abi ja tuge. Kuna vaimsed võimed on piiratud, tuleb arvestada lapse arenguga. Võrdlus samaealiste mängukaaslastega pole lubatud. Õppeprotsess peaks olema kohandatud ka patsiendi võimetele ja oskustele.
Haiguse käigus toimub sageli esimestel eluaastatel mitu kirurgilist sekkumist. Nende väljakutsetega toimetulemiseks tuleks organismi toetada tervisliku, vitamiinirikka toitumisega. Optimaalne unehügieen ja stressi vältimine aitavad ka sümptomeid leevendada. Lisaks väärarengutele kannatavad paljud patsiendid ka kuulmislanguse all. Seda asjaolu tuleb õnnetuste ja keeleoskuse vähendamisel igapäevaelus arvestada. Sihipärane koolitus aitab parandada keeleoskust ja suhtlust teiste inimestega.

.jpg)