Mowat-Wilsoni sündroom on haruldane geneetiline arenguhäire, millel on lai valik sümptomeid. Lisaks näo-, soole- ja suguelundite anomaaliatele esinevad geneetilise defekti osana ka südamedefektid ja aju arenguhäired. Seni ravimatut haigust saab ravida ainult sümptomaatiliselt.
Mis on Mowat-Wilsoni sündroom?

© Jezper - stock.adobe.com
Mowat-Wilsoni sündroom on üsna noor kliiniline pilt. Kliiniliselt mitmekesist nähtust kirjeldasid Mowat ja Wilson esmakordselt 1998. aastal. Lisaks arenguhäiretele iseloomustavad kliinilist pilti mikrotsefaalia ja Hirschsprungi haiguse sümptomikompleks. Haiguse põhjustajaks peetakse geneetilist defekti.
Üldiselt on sümptomid äärmiselt mitmekesised. Haruldast haigust on seni vähe uuritud. Selle tulemusel on seni saadaval vähe ravivõimalusi. Lõplikku levimust ei ole, kuna 21. sajandil võis häireid diagnoosida harva või üldse mitte. Praegu on sündroomiga umbes 200 dokumenteeritud patsienti.
põhjused
Geenimutatsioon põhjustab Mowat-Wilsoni sündroomi. Värskete uuringute kohaselt on haigust põhjustav geen ZFHX1B. Väidetavalt on põhjuslik geneetiline defekt kromosoomi piirkonnas 2q22. Mõjutatud geen on umbes 70 kb suurune ja koosneb kokku kümnest 1214 aminohappe eksonist. See geen kodeerib valku SIP1, mis on aktiivne transkriptsiooni modulaatorina ja osaleb embrüogeneesis.
Seetõttu on mõjutatud inimeste embrüogenees häiritud. Geeni haigust põhjustavad anomaaliad võivad vastata täielikule deletsioonile, ümberpaigutamisele või järjestikusele anomaaliale. Geneetiline defekt kandub edasi autosomaalses domineerivas pärandis. Päriliku haiguse edasiandmiseks piisab kahe homoloogse kromosoomi puudulikust alleelist.
Sümptomid, tervisehäired ja nähud
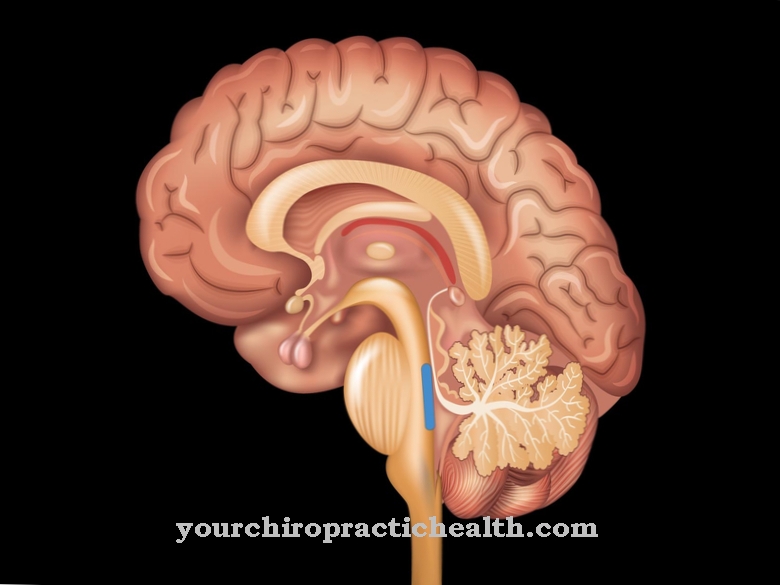
Mowat-Wilsoni sündroomi sümptomid vastavad keerukale arenguhäirele ja on kliiniliselt mitmekesised. Peamisteks sümptomiteks on tserebraalselt vallandunud krambid ja mikrotsefaalia. Selline mikrotsefaalia tekib kõigi kraniaalsete õmbluste enneaegse kõvenemise tagajärjel ja ahendab kasvufaasis aju. Seetõttu kogevad patsiendid vaimset alaarengut. Lisaks on sageli näo kõrvalekaldeid, mis sageli annavad patsiendile kotkakujulise profiili.
Need kõrvalekalded võivad hõlmata näiteks suuri, sügavalt asetsevaid silmi, horisontaalselt suunatud kulme, aurikli kõrvalekaldeid, sissekasvanud kõrvakellasid ja silmatorkavalt väljaulatuvat lõua. 90 protsendil juhtudest kannatavad epilepsia all kannatajad. Vaimne areng on tugevalt hilinenud ja keeleline areng puudub täielikult. Samuti aeglustub patsiendi motoorne areng.
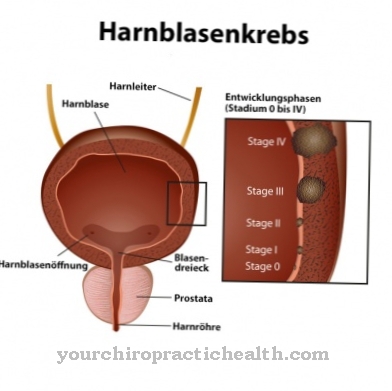
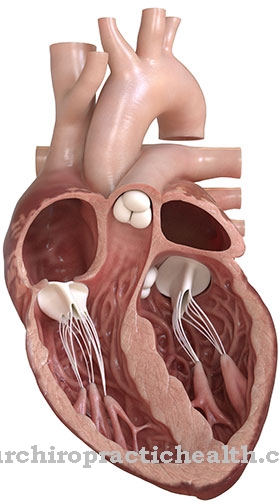
Normaalsete sünnimõõtmiste korral esineb sageli sekundaarset lühikest kasvu. Võib esineda ureetra väärarenguid. Mõeldavad on ka kaasasündinud südamedefektid või suguelundite väärarengud. Lisaks ilmnevad soole seina plexuse neuronaalsed kõrvalekalded, kuna need on iseloomulikud Hirschsprungi tõvele.
Diagnoos ja haiguse kulg
Mowat-Wilsoni sündroomi ei saa diagnoosida pelgalt uuringute põhjal, vaid see nõuab geneetilise materjali analüüsi. Labor amplifitseerib patsiendi genoomsest DNAst ZFHX1B geeni kaks kuni kümme eksonit. See amplifikatsioon toimub PCR abil. Selle materjali ja introni-eksoni splaissimiskohtade analüüs toimub DNA sekveneerimise abil.
Iga ZFHX1B geeni eksonit uuritakse deletsiooni ja dubleerimise suhtes multipleksse ligatsiooni sõltuva sondi amplifikatsiooni kaudu. See põhjalik protseduur võtab aega umbes kolm nädalat ja erinevalt patsiendi lihtsalt läbivaatusest võimaldab see teha ühemõttelise diagnoosi. Enamasti sekveneeritakse ja analüüsitakse lisaks inimese DNA-le ka tema vanemate DNA-d.
Haiguse käik sõltub suuresti geneetilise kõrvalekalde vormist ja kromosoomi osade kustutamise või ümberpaigutamise ulatusest. Siiani väheste dokumenteeritud haigusjuhtumite tõttu pole lõplikke prognoose võimalik teha. Kuid varajane diagnoosimine ja sellele järgnev ravi mõjutavad prognoosi tõenäoliselt positiivselt.
Tüsistused
Mowat-Wilsoni sündroom põhjustab patsiendil tõsiseid kaebusi ja tüsistusi, mis vähendavad märkimisväärselt eeldatavat eluiga ja elukvaliteeti. Reeglina on patsiendi igapäevane elu ka märkimisväärselt piiratud ja mõjutatud inimesed sõltuvad igapäevaelus teiste inimeste abist.
Lisaks on vaimne alaareng, kus sugulased ja vanemad kannatavad sageli psühholoogiliste kaebuste või depressiooni all. Enamasti kannatavad kannatanud isikud ka krambi ja vähenenud vastupanuvõime all. Lisaks ilmnevad ka mitmesugused näo deformatsioonid ja epilepsia.
Keeleline areng lükkub samuti märkimisväärselt edasi, nii et täiskasvanueas on patsiendiga suhtlemisel suuri raskusi. Esineb ka südamepuudulikkust ja lühikest kasvu. Südamedefekt võib põhjustada südame spontaanse surma, nii et Mowat-Wilsoni sündroom piirab haigestunud inimese eluiga.
Mowat-Wilsoni sündroomi ei saa ravida. Erinevaid kaebusi saab siiski piirata ja käsitleda nii, et asjassepuutuval inimesel oleks igapäevaelu talutav. Komplikatsioone pole, kuid positiivne ravi pole alati võimalik.
Millal peaksite arsti juurde minema?
Ehkki Mowat-Wilsoni sündroomi ei saa praeguste juriidiliste ja meditsiiniliste võimalustega ravida, võib tekkivate sümptomite ravi pakkuda olulist leevendust. Tavaliselt, mida varem diagnoosi saab panna, seda paremad on patsiendi ravivõimalused. Kui kasvaval lapsel ilmnevad arenguhäired, on vajalik arstiga konsulteerimine.
Kui otseses võrdluses samaealistega on individuaalseid kõrvalekaldeid, on vajalik arst. Vaatlusi tuleks temaga arutada, et oleks võimalik hinnata tervislikku seisundit. Arst peaks olema tutvustatud õpiraskuste, mäluhäirete, kõne hilinemise või liikumisjärjestuste iseärasustega. Kui teil tekivad krambid, valu või ebaharilik rüht, peate nägema arsti. Näo väärarengud või kõrvalekalded näitavad seisundit, mis vajab ravi.
Nägemisvigu või näojoonte kõrvalekaldeid peab arst selgitama. Aeglustatud mõtteprotsessid või liikumised on häire tunnused ja neid tuleks uurida. Südame rütmihäirete, väljaheidete probleemide või reageerimise või tajumise võime ebakorrapärasuse korral tuleb pöörduda arsti poole. Käitumishäireid, vegetatiivseid häireid või naha väljanägemise iseärasusi peab uurima arst.
Ravi ja teraapia
Mowat-Wilsoni sündroom on siiani ravimatu. Samuti on piiratud sümptomaatilise ravi võimalused. Krampide vastu kasutatakse tavaliselt ravimeetodeid. Epilepsiavastased ravimid näitavad selles kontekstis suurimat efektiivsust. Mõnda sümptomaatilist väärarengut saab korrigeerida kirurgiliselt. Eelkõige tuleks võimalikult varakult korrigeerida Hirschsprungi tõve sümptomeid, sest vastasel juhul võib tekkida sepsis või peritoniit.
Mowat-Wilsoni sündroomi sümptomaatiline ravi on peamiselt ette nähtud kannatanute elukvaliteedi parandamiseks. Selle eesmärgi saavutamiseks saab võidelda ka vaimse ja motoorse alaarenguga. Logopeedilised teraapiad võivad teatud olukordades aidata keele arengut, mis Mowat-Wilsoni sündroomi korral ebaõnnestub sageli ilma toetavate terapeutiliste abinõudeta. Füsioterapeutiline ja tegevusteraapia ravi võib motoorsete oskuste hilinenud arengule vastu tulla.
Mowat-Wilsoni sündroom on haigestunud inimese vanematele sageli peaaegu kujuteldamatu psühholoogiline koormus. Sel põhjusel toetavad patsientide vanemaid sageli psühhoterapeudid. Meditsiinilised uuringud on seotud geeniteraapia lähenemisviisidega, mis peaksid tulevikus parandama geenipuudujääke. Sel viisil saab kahjustatud ZFHX1B geen varsti asendada, mis võib muuta haiguse ravitavaks.
Outlook ja prognoos
Mowat-Wilsoni sündroomi saab tänapäeval hästi ravida. Eeldatava eluea ja elukvaliteedi aluseks on kaasasündinud väärarengute tüüp ja raskusaste. Kergete kõrvalekallete korral, mis ei mõjuta südant, võivad haigestunud inimesed elada täiskasvanueas.
Tõsiselt haiged patsiendid surevad haiguse tagajärjel tavaliselt lapsepõlves või noorukieas. Tüüpilised surmapõhjused on müokardiinfarkt või iseloomulikud HSCR-haigused. Ajuhoogude tagajärg on lapse esimestel eluaastatel sageli surm. Haruldast sündroomi saab ravida sümptomaatiliselt, mis tähendab, et patsiendid võivad vähemalt ajutiselt elada sümptomiteta elu.
Pikemas perspektiivis ei paku Mowat-Wilsoni sündroom siiski positiivset prognoosi, kuna mitmesugused väärarengud ja kõrvalekalded põhjustavad tervise järkjärgulist halvenemist ja lõppedes surmaga. Eeldatava eluea ja haiguse kulgu prognoosib tavaliselt vastutav spetsialist. Enamasti on see neuroloog või geneetiliste haiguste spetsialist. Sõltuvalt sümptomitest võib haiguse diagnoosimine olla keeruline, mistõttu ei diagnoosita sageli Mowat-Wilsoni sündroomi enne, kui haigus on kaugele arenenud.
ärahoidmine
Kuna Mowat-Wilsoni sündroom on keeruline arenguhäire, millel on geneetiline põhjus, saab seda nähtust vaevalt ennetada. Pereplaneerimisega seotud paaridel võib siiski olla DNA järjestus, et hinnata nende isiklikku riski geneetiliste defektide edasikandumiseks.
Järelhooldus
Enamikul juhtudest ei ole Mowat-Wilsoni sündroomiga nakatunud patsientidel järelmeetmeid või on neid vähe, kuna tegemist on geneetilise haigusega. Seetõttu peaksid kannatanud isikud ideaaljuhul konsulteerima arstiga varakult, et ei tekiks täiendavaid kaebusi ega komplikatsioone, millel võib olla negatiivne mõju asjaomase inimese eeldatavale elueale ja elukvaliteedile.
Reeglina ei saa iseenesest paraneda, nii et haiguse esimeste nähtude ja sümptomite ilmnemisel tuleb pöörduda arsti poole. Kui soovite lapsi saada, võib geneetiline testimine ja nõustamine olla kasulik sündroomi kordumise vältimiseks teie järeltulijatel. Reeglina sõltuvad Mowat-Wilsoni sündroomist mõjutatud isikud mitmesugustest ravimitest.
Neid tuleb sümptomite leevendamiseks alati võtta õigeaegselt ja õigetes annustes. Laste puhul peaksid tarbimist kontrollima eelkõige vanemad. Füsioteraapia meetmed on samuti paljudel juhtudel vajalikud, ehkki osa harjutusi saab läbi viia ka oma kodus. Ei saa üldiselt ennustada, kas Mowat-Wilsoni sündroom viib mõjutatud inimese eluea lühenemiseni.
Saate seda ise teha
Kuna Mowat-Wilsoni sündroomi paraku ei ravi, on praegu peamine prioriteet lapse elukvaliteedi parandamine.
Paljudel juhtudel võib varakult alustatud kõneteraapia tasakaalustada viivitatud keelelist arengut ja tagada keele arengule märkimisväärse edu. Lisaks tagavad intensiivsed füsioterapeutilised ja tegevusteraapia abinõud parema motoorse ja vaimse arengu. Lisaks arsti määratud abinõudele on soovitatav teemaga ise tegeleda ja ravi jätkata kodus.
Puudega lapse eest hoolitsemine on tohutu koorem, eriti vanemate jaoks, aga ka kõigi võimalike õdede-vendade jaoks, kes võivad mõjutada pereelu ja lõppkokkuvõttes hoolduse kvaliteeti. Seetõttu on äärmiselt oluline, et sellistel puhkudel pöörduksid vanemad õigeaegselt psühhoteraapia poole, mis annab neile pikemas perspektiivis rohkem jõudu, õppides lõõgastumise ja konfliktide lahendamise meetodeid.
Samuti tuleb meeles pidada, et kannatanud isikutel on igal aastal õigus kuni kuue nädala pikkusele ennetavale hooldusele, mille kulud katab hoolduskindlustus. Juba on olemas rajatisi, mis pakuvad intensiivravi päevasel ajal, samal ajal kui sugulased saavad ekskursioonidel puhata. Sellest võib olla palju abi, eriti õdede-vendade puhul.