Muenke sündroom iseloomustab pärgarteri õmbluse kraniosüostoos, mis on tingitud FGFR3 geeni mutatsioonist. Häire on päritav autosomaalse domineeriva tunnusena ja sellega kaasnevad sageli ebanormaalsed jäsemed. Ravi vastab tavaliselt kirurgilisele protseduurile.
Mis on Muenke sündroom?

© flashmovie - stock.adobe.com
Kraniosünostoosi korral luustub üks või mitu koljuõmblust embrüonaalse arengu ajal enneaegselt ja takistavad sellega kolju ja aju füsioloogilist kasvu. Paljud haigused, mis kuuluvad kaasasündinud väärarengute sündroomide rühma, kus esinevad peamiselt näod, sisaldavad selliseid kraniosüostoose. Selline haigus see on Koronaarõmbluse sünostoosi sündroom, nimetatud ka Muenke sündroom on tuntud.
Haigust kirjeldati esmakordselt 1997. aastal. M. Muenke ja tema kolleegid kirjeldavad seda esimest korda. Muenke sündroomi iseloomustab pärgarteri õmbluse kraniosinostoos ja see hõlmab ka skeleti muutusi tarsaalses ja karpaalis. Sündroomi levimus pole praegu teada. Manifestatsioon toimub varajases vastsündinu perioodil või hiljemalt väikelapse varases eas. Kuigi haigust ei ole lõplikult uuritud, on selle põhjus nüüdseks selgitatud.
põhjused
Paljudel juhtudel võis Muenke sündroomi seostada perekondliku kuhjumisega. Nendel juhtudel sarnaneb pärand kõige otsesemalt autosomaalse domineeriva pärandiga. Siiski on dokumenteeritud ka juhtumeid, kus sündroom näis ilmnevat juhuslikult. Põhjus näib olevat geneetiline mutatsioon, mis sporaadilistel juhtudel vastab tõenäoliselt uuele mutatsioonile. Mutatsiooni asukohta peetakse samuti tuvastatuks.
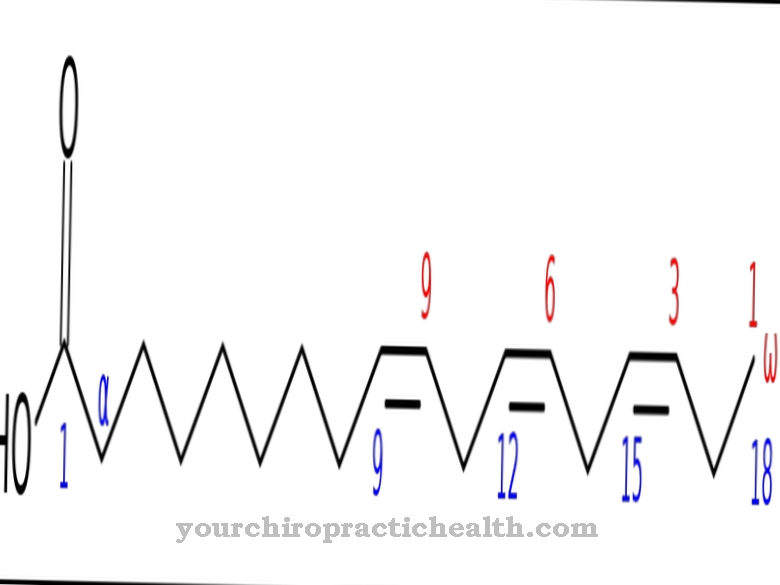
Väidetavalt põhineb haigus FGFR3 geeni mutatsioonidel, mida võib leida geeni lookuses 4p16.3. FGFR3 geeniga on seotud ka muud sündroomid. Selle üheks näiteks on nn Aperti sündroom. Geen kodeerib DNA sees fibroblastide kasvufaktori retseptori 3. FGF-3 kasvufaktori füsioloogiliste mõjude kohta on vähe teada.
Spekulatsioonide kohaselt on FGF-3 eriti embrüonaalse perioodi jaoks otsustav tegur. Retseptorite mutatsioon tähendab eeldatavalt seda, et kasvufaktor ei seo embrüonaalse arengu ajal piisavalt.
Sümptomid, tervisehäired ja nähud
Muenke sündroomiga patsientidel on mitmesuguseid sümptomeid. Koronaarsete õmbluste enneaegse sulgemise tõttu on mõjutatud isikutel ebanormaalne pea kuju, mis on märgatav ka näo anomaaliates. Lisaks lühendatud kolju eesmisele ja tagumisele läbimõõdule on silmade pistikupesad tavaliselt vähem sügavad.
Sageli on need sümptomid seotud ülemise lõualuu hüpoplaasiaga. Kui pärgarteri õmblus on ühelt poolt suletud, on silmade pistikupesad vastaval küljel lamestatud. Tavaliselt ei mõjuta sündroom patsiendi intelligentsust. Jäsemetel leitakse kätel või tarsal luud. Valegregatsioon on mõeldav ka karpaalluudel.
Koonu epifüüsid on ka üks võimalikest sümptomitest. Mõnel juhul on patsiendi kliiniline pilt seotud ka osteokondroomidega. Fenotüüpne ja seega sümptomaatiline kattumine teiste sündroomidega nagu Pfeifferi sündroom, Jackson-Weissi sündroom või Saethre-Chotzeni sündroom on kliiniliselt mõeldavad ilmingud.
Diagnoos ja haiguse kulg
Muenke sündroomi diagnoos tehakse tavaliselt vastsündinu perioodil, kuna visuaalse diagnoosi abil saab haiguse varakult ära tunda. Umbes üks patsient 15 000 vastsündinust kannatab pärgarteri õmbluse sünostoosi all. See nähtus ei pea aga automaatselt tulema Muenke sündroomi tõttu.
Seetõttu nõuab diagnostika FGFR3 geeni patogeense mutatsiooni tuvastamist. Patsiendi käed ja jalad võivad radioloogiliselt normaalsed tunduda, nii et diagnoosi saamiseks ei piisa selles osas kõrvalekallete otsimisest. Põhimõtteliselt saab kõiki pärgarteri sünostoosiga lapsi skriinida spetsiifilise P250R mutatsiooni suhtes.
See uurimine vastab molekulaargeneetilisele analüüsile. Mutatsiooni välistamine ei tähenda tingimata, et patsient ei põeks Muenke sündroomi. Mõnel juhul ei olnud mutatsiooni tuvastatud haigetel. Tõendit peetakse siiski diagnoosi kinnitamiseks. Naispatsientide prognoos on ebasoodsam.
Tüsistused
Muenke sündroomi tõttu kannatavad mõjutatud mitmesugused väärarengud ja deformatsioonid, mis esinevad peamiselt patsiendi peas ja näos. Need deformatsioonid põhjustavad sageli psühholoogilisi kaebusi ja depressiooni. Mõjutatud kannatavad sageli alaväärsuskomplekside ja madalama enesehinnangu all.
Sümptomid põhjustavad sageli häbi ja eriti lapsi võib Muenke sündroomi tõttu kiusamine ja kiusamine mõjutada. Kuid intelligentsus ei ole halvenenud, nii et patsiendi vaimne areng kulgeb komplikatsioonideta. Samuti on jäsemetes väärarenguid, nii et igapäevaelus võivad olla mitmesugused tegevused või liikumispiirangud.
Elukvaliteeti vähendab märkimisväärselt Muenke sündroom. Muenke sündroomi ravis pole muid tüsistusi. Põhjuslik ravi ei ole reeglina võimalik, kuid edaspidiste tagajärjetu kahjustuste vältimiseks tuleb läbi viia mõned kirurgilised sekkumised.
Asjaomase isiku eluiga tavaliselt ei piirata. Ravi võib toimuda ka kohe pärast sündi. Mõnel juhul mõjutavad Muenke sündroomi psühholoogilised kaebused ka laste vanemaid.
Millal peaksite arsti juurde minema?
Pea ja näo iseloomulikud väärarengud näitavad Muenke sündroomi selgelt ja viivad diagnoosi tavaliselt kohe pärast sündi. Kui sümptomid on kerged, tuleb vastutavat arsti kõigist sümptomitest teavitada. Mõnikord on karpaal- või tarsaalluud kokku kasvanud, mille tulemuseks on sellele haigusele tüüpiline kõnnak. Inimestel, kellel on juba haiguse perekondlik ajalugu, tuleks geneetiline test teha varases staadiumis. Seejärel saab kohe pärast sündi alustada vajalikke ravitoiminguid.
Haigestunud laste vanemad peaksid oma perearsti teavitama ka lapse uutest sümptomitest ja muust kalduvusest. Lisaks on alati vajalik spetsialisti põhjalik kontroll. Ravi toimub tavaliselt haiglas või geneetiliste haiguste kliinikus. Lisaks üldarstile saab sisse kutsuda ortopeedi või sisearsti. Krooniliste kaebuste korral on ravi osa ka teraapiast ja füsioteraapiast.
Teraapia ja ravi
Muenke sündroomiga patsientidel pole põhjuslikku ravi võimalik. Geeniteraapia lähenemisviisid pakuvad lootust põhjuslikule teraapiale, kuid need pole veel kliinilisse faasi jõudnud. Ravi on puhtalt sümptomaatiline ja sõltub seega konkreetse juhtumi sümptomitest. Kolju kõrvalekallete parandamiseks saab kasutada ainult kirurgilisi ravivõimalusi.
Kirurgiline protseduur on ette nähtud ajusurve vähendamiseks, mis on põhjustatud pärgarteri õmbluse varasest sulgemisest. Kraniaalnärvid vabastatakse sel viisil ja ideaaljuhul on need protseduuri järgselt vähem või üldse mitte kokku surutud. Kergemate kraniosünostooside korral on saadaval konservatiivne ravivõimalus. Näiteks võib kergemini mõjutatud lastele anda kolju kuju, mida nad peavad pikka aega kandma.
Need koljuvormid üritavad kolju adekvaatselt modelleerida. Kuna Muenke sündroomi diagnoositakse tavaliselt juba vastsündinul, on selline konservatiivne ümberkujundamine eriti kasulik: väikelaste pea kuju on endiselt kohanemisvõimeline. Konservatiivsel modelleerimisel on lõppkokkuvõttes sama eesmärk nagu operatiivsel modelleerimisel.
Ravi peaks võimaldama aju füsioloogilist kasvu. Lisaks kohandatakse pea välimust keskmisele. Samaaegseid sümptomeid, nagu jäsemete väärarengud, saab ravida kirurgiliselt. Kui need ei piira ega takista mõjutatud isikut, pole selline ravi tingimata vajalik.
Outlook ja prognoos
Muenke sündroom on haruldane haigus, mida saab nüüd ravida kirurgiliselt. Kui pärgarteri õmbluse enneaegne sulgemine tuvastatakse õigeaegselt, on prognoos hea. Kõiki väärarenguid saab ravida kirurgiliselt. Selliste sümptomite jaoks nagu valu või tundlikkuse häired võib välja kirjutada sobivad ravimid. Pärgarteri õmbluste sünostoosi sündroom ei vähenda eeldatavat eluiga.
Elukvaliteeti võivad pisut piirata nii mainitud kaebused kui ka võimalikud armid näol ja peas. Üldiselt on prognoos siiski positiivne. Muenke sündroomiga lapsed on eriti altid sisekõrva kuulmislangusele. Halb kuulmine võib põhjustada probleeme hilisemas elus, näiteks juhul, kui patsient ei suuda enam juhistest aru saada ega oma eluteed igapäevaelus leida.
Tõrjutuse ja vähenenud enesehinnangu tõttu tekivad mõnel haigel psühholoogilised kaebused. Heaolu saab vähendada, kui haigetel puudub hea tugivõrgustik vanemate, sugulaste, sõprade ja terapeutide näol. Muenke sündroomi all kannatavad lapsed vajavad igapäevaelus kindlasti tuge, et jääda vaimu ja füüsiliselt terveks hoolimata haigusest.
ärahoidmine
Muenke sündroomi ei saa vältida, kuna seda mõjutavad pigem geneetilised, mitte välised tegurid. Ainus ennetav meede on raseduse ajal nõustatav geneetiline nõustamine.
Järelhooldus
Enamikul juhtudel on Muenke sündroomiga nakatunutel otseseks järelraviks väga vähe ja ainult väga piiratud meetmeid. Kõigepealt tuleks selle haiguse kiire ja ennekõike varajane diagnoosimine teha nii, et edasised komplikatsioonid ei tekiks. Kuna Muenke sündroom on geneetiline haigus, ei saa seda täielikult ravida.
Kui aga soovite lapsi saada, võib sündroomi kordumise vältimiseks olla kasulik geneetiline testimine ja nõustamine. Enamik kannatanutest sõltub operatsioonist. Patsient peaks pärast sellist operatsiooni kindlasti puhata ja rahulikult võtma, ideaaljuhul tuleks hoida voodipuhkust. Samuti ei tohiks keha asjatult koormata, vaid ka pingelist või füüsilist tegevust.
Paljudel juhtudel sõltuvad sündroomi all kannatajad ka oma pere abist ja toetusest igapäevaelus. Armastavad ja intensiivsed vestlused pere ja sõpradega mõjutavad ka haiguse edasist käiku väga positiivselt. Pole sugugi haruldane, et vältida depressiooni või muude psühholoogiliste häirete teket.
Saate seda ise teha
Muenke sündroomi ravitakse tavaliselt kirurgiliselt ja ravimitega. Kõige olulisem eneseabimeede on hoolitseda keha eest pärast protseduuri ja konsulteerida tihedalt vastutava arstiga. Mõjutatud laste vanemad peaksid last tähelepanelikult jälgima ja kõrvalekallete ilmnemisel teavitama vastutavat arsti.
Üldiselt saab Muenke sündroomi suhteliselt hästi ravida ega põhjusta püsivat halvenemist elukvaliteedis. Deformatsioonid ja väärarengud jäävad aga peaaegu alati alles, mis võib mõnikord olla kannatanutele suur emotsionaalne koormus. Sel põhjusel tuleks meditsiinilist ravi toetada terapeutiliste meetmetega.
Arenguhäiretega tõsiselt kannatanud lastele soovitati varem käia spetsiaalses lasteaias ja hiljem erikoolis. Selle asemel viidatakse täna tavakoolidesse kaasamisele. Muude kõrvalekalleteta lapsed saavad nagunii minna tava- ja kõrgkooli.
Põhjalik ravi on vajalik, eriti raskete liikuvuspiirangute korral. Vanemad saavad neid abinõusid toetada, toetades last igapäevaelus. Tavaliselt on näidustatud täiendavad füsioterapeutilised meetmed.
Kuna haigus võib olla ka märkimisväärne koormus sugulastele, peaksid vanemad ja sõbrad otsima ka terapeutilist abi. Terapeut saab luua kontakti ka teiste haigestunud isikutega ja vajadusel suunata vanemad eneseabigruppi.
.jpg)

.jpg)