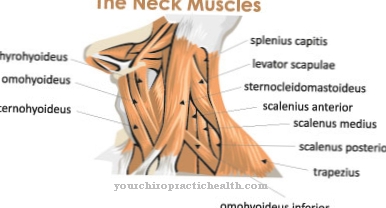
Ensüümi asendusravi kasutatakse lüsosomaalsete säilitushaiguste raviks, mille korral ensüümide puudus põhjustab lagunemisproduktide patoloogilist kogunemist rakkude lüsosoomidesse.
Geneetiliste defektide tõttu puuduvad ensüümid kompenseeritakse regulaarsete intravenoossete infusioonidega. Kuna infundeeritud sünteetilised ensüümid ei saa oma molekulaarse suuruse tõttu ületada hematoentsefaalbarjääri, toimib ravi ainult lüsosomaalsete säilitushaiguste korral, mis ei mõjuta kesknärvisüsteemi.
Mis on ensüümi asendusravi?

Lüsosoomid on spetsiaalsed rakuorgaanid, milles võõr- ja endogeensed ained lagundatakse ja osaliselt taaskasutatakse. Ainete lagundamiseks ja transportimiseks on vaja spetsiifilisi hüdrolüüsivaid ensüüme. Need on proteaasid, nukleaasid, lipaasid ja transporterid.
Mitmed teadaolevad geneetilised defektid võivad põhjustada teatud ensüümide tõrkeid, nii et mõned laguproduktid kogunevad lüsosoomidesse patoloogilistes kogustes ja akumuleeruvad seni, kuni nad jõuavad kontrollimata viisil rakuvälisesse maatriksisse, st rakudevahelistesse ruumidesse. Kõik geneetilised vead, mis põhjustavad vähemalt ühe vajaliku hüdrolaasi rikke, on kokku võetud terminiga lüsosomaalne säilitushaigus. Ensüümi asendusravi (ERT, ensüümi asendusravi) kasutatakse puuduvate endogeensete ensüümide asendamiseks sünteetiliselt toodetud ensüümidega.
Kuna hüdrolaasid koosnevad suhteliselt suurtest molekulidest, ei saa nad soolest imenduda, enne kui need on lagundatud ja inaktiveeritud, nii et neid saab manustada ainult veenisisese infusiooni teel. Ensüümimolekulide suurus takistab aga ka vere-aju barjääri ületamist, nii et teraapia saab olla efektiivne ainult lüsosomaalsete säilitushaiguste korral, mis ei mõjuta kesknärvisüsteemi (KNS).
Funktsioon, mõju ja eesmärgid
On teada üle 50 erineva lüsosomaalse ainevahetushäire, millest igaühe põhjustajaks on monogeneetiline defekt. Lüsosomaalsed säilitushaigused võib jagada seitsmesse erinevasse klassi sõltuvalt olemasoleva ensüümi puuduse tõttu ülemääraselt ladustatud ainetest.
Mukopolüsahhariidid ja oligosahhariidid sobivad peamiselt ERT-le. ERT eesmärk on alati kompenseerida spetsiifiliste ensüümide puudus kunstlikult tarnitud ensüümide abil, et viia haigus seisma või vähemalt leebemale kulgemisele. Täpsemalt on asendusensüümid saadaval järgmiste lüsosomaalsete säilitushaiguste korral:
- Gaucheri tõbi
- Pompe tõbi
- Fabry haigus
- Hurleri-Pfaundleri sündroom (mukopolüsahharoos I)
- Jahimehe tõbi (mukopolüsahharoos II)
• Maroteaux-Lamy sündroom (mukopolüsahharoos VI) • Niemann-Pick B
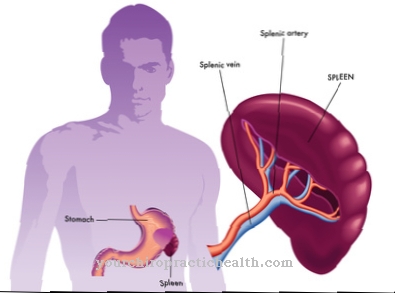
Gaucheri tõbi on kõige levinum lüsosoomide säilitushaigus. Seda esineb kolmes erinevas variandis, millest kaks mõjutavad ka närvisüsteemi. Mitteneuropaatilises vormis mõjutab eriti põrn, mis suureneb oluliselt ja põhjustab sekundaarseid kahjustusi nagu aneemia ja luuüdi kahjustused. Tüüpilised sümptomid on luu- ja liigesevalu ning vereringehäired. Haiguse äge neuropaatiline variant näitab rasket kulgu ja pakub väheseid võimalusi ellujäämiseks kahel esimesel eluaastal.
Ladustamishaigus Pompe tõbi on põhjustatud ensüümi alfa-1,4-glükosidaasi puudulikkusest, mis osaleb paljudes metaboolsetes protsessides. Pompe tõbi põhjustab südame tohutut laienemist (kardiomegaalia) ja südamepuudulikkust. On varasemaid, tõsiseid kursusi, mis ilmuvad esimestel elukuudel, aga ka leebemaid vorme, mis ilmuvad alles hilisematel eluaastatel.
Fabry haiguse põhjustab X-seotud geneetiline defekt, seega võib säilitushaigus mõjutada ainult poisse ja mehi. Haigus põhjustab tavaliselt kaugelearenenud lapseeas sümptomeid, sealhulgas valuhooge, naha keratoome, neeruprobleeme ja südamelihase kahjustusi. Ensüümi alfa-galaktosidaas A defitsiit põhjustab tseramiidtriheksoosiidi kuhjumist, mis on sümptomite esilekutsumise põhjus, mis võib mõjutada ka autonoomset närvisüsteemi.
Pole harvad juhud, kui kahjustus põhjustab südameinfarkti, neeruinfarkti või isegi insuldi. Hurleri-Pfaundleri sündroomi nimetatakse ka I tüüpi mukopolüsahharoosiks ja see on põhjustatud glükosaminoglükaani metabolismi häiretest. Haigus on seotud mitmesuguste sümptomitega, sealhulgas raske vaimse kahjustuse ja luustiku tõsiste muutustega. Haiguse kulg on raske, nii et keskmine eluiga on 11–14 aastat. Hunteri tõbi vastab 2. tüüpi mukopolüsahharoosile ja on - nagu ka Hurleri tõbi - põhjustatud X-seotud defektist. Haigust iseloomustavad erineva raskusastmega kursused, mis esinevad varases lapsepõlves kuni kergete kuurideni, mis ilmnevad ainult täiskasvanud meestel.
Kõige tavalisemate südame sümptomite, näiteks südameklappide defektide ja südamelihase probleemide tõttu varieerub oodatav eluiga normaalsest kuni pisut piiratud. Maroteaux-Lamy sündroom (MPS VI) on üks mukopolüsahhariidoosidest, mis päritakse autosomaalselt retsessiivselt, kuna põhjuslikku geeni puudust ei ole X-kromosoomis. Haigus on väga haruldane, üks juhtum 455 000 sündi kohta. On teada kerged ja rasked vormid.
Sümptomiteks on suurenenud maks ja põrn, karpaalkanali sündroom ja muutused südameklappides. Niemann-Pick B on sfingomüeliini lipidoos, mis on üks lüsosoomide säilitushaigustest ja põhjustatud 11. kromosoomi geneetilisest defektist. Kui haiguse tüüp B mõjutab peamiselt maksa ja põrna, on A-tüübil ka märkimisväärseid neuroniprobleeme.
Ravimid leiate siit
➔ ValuravimidRiskid, kõrvaltoimed ja ohud
Kuna paljud lüsosoomide säilitushaigused, mida saab ravida ensüümi asendusraviga, läbivad raske kursuse ja vastavalt suureneb suremus, on ERT-s suurim oht, et valitud asendusensüüm ei tööta või töötab ainult liiga nõrgalt.
Teine oht peitub vähem teraapias kui selles, et põhihaigust tuvastatakse liiga hilja, nii et ERT võib kuuri jooksul peatuda, kuid juba tekitatud kahju ei saa taanduda. Umbes iga teine ravitav patsient reageerib infusioonile ajutiselt selliste sümptomitega nagu palavik ja külmavärinad. Selle põhjused pole veel täielikult teada. Mõned patsiendid reageerivad antikehade moodustumisega ja on teada juhtumeid, kus patsiendid on reageerinud löövete ja bronhospasmiga.





.jpg)








.jpg)
.jpg)